 Elektoforeza Kapilarna
Elektoforeza Kapilarna
Na podstawie artykułu dr Ewy Poboży “Elektroforeza kapilarna” opublikowanego w kwartalniku ANALITYKA Nr 2/2001
Elektroforeza kapilarna – fascynujące narzędzie w rękach analityka
Jednym z ważnych zadań chemii analitycznej jest opracowywanie metod umożliwiających jakościową i ilościową analizę próbek wieloskładnikowych. Ze względu na brak wysoce selektywnych metod detekcji, niejednokrotnie konieczne jest wstępne rozdzielanie oznaczanych analitów. Efektem poszukiwań odpowiednich metod analitycznych jest rozwój różnorodnych technik analizy instrumentalnej.
Obecnie analitycy dysponują nowym narzędziem – wysoko sprawną elektroforezą kapilarną, metodą, która w ostatnich latach cieszy się ogromnym zainteresowaniem. Ze względu na procesy które decydują o rozdzielaniu, sposób prowadzenia pomiarów, a także możliwości, jakie stwarza, jest to metoda naprawdę fascynująca. Zjawisko elektroforezy, czyli migracji cząstek obdarzonych ładunkiem w polu elektrycznym w kierunku elektrody o przeciwnym znaku, znane było już pod koniec XIX wieku. Pierwszym, który zastosował w 1937 roku elektroforezę jako metodę rozdzielania, był A. W. Tiselius, a jego prace, dotyczące rozdzielenia białek, uhonorowane zostały Nagrodą Nobla. Początkowo eksperymenty prowadzone były w roztworach buforów w specjalnych U-rurkach. Jednak ze względu na znaczną konwekcję i dyfuzję, spowodowaną wydzielaniem się ciepła, osiągano małe sprawności rozdzielania. Dopiero zastosowanie stabilizujących wypełnień, takich jak: agar, żel poliakryloamidowy, proszek celulozowy, wata szklana pozwoliło zmniejszyć niekorzystne efekty. Do dzisiaj elektroforeza planarna bibułowa i żelowa jest często stosowana do rozdzielania biomolekuł.
Ograniczenie konwekcji i dyfuzji możliwe było również dzięki zastosowaniu rurek o małych średnicach, w których prowadzono rozdzielanie. Prawdziwy przełom w rozwoju elektroforezy kapilarnej nastąpił w 1981 roku, kiedy ukazały się prace Jorgensona i Lukacsa. Datę tę uznaje się za początek wysoko sprawnej elektroforezy kapilarnej (HPCE). Zastosowanie kapilar o średnicach 75 µm znacznie ograniczyło wpływ wydzielanego ciepła nawet przy stosowaniu napięcia rzędu 30 kV i pozwoliło uzyskać wysokie sprawności. W kolejnych latach nastąpił dynamiczny rozwój metody w zakresie teorii oraz zastosowania jako metody analitycznej i aparatury pomiarowej. Mimo że HPCE jest stosunkowo młodą metodą, to w obecnej formie umożliwia bardzo dokładne jakościowe i ilościowe oznaczenia analityczne. Dzięki swoim zaletom ta nowoczesna metoda znalazła szerokie zastosowanie i uznawana jest jako alternatywna lub komplementarna metoda analityczna w stosunku do metod chromatograficznych.
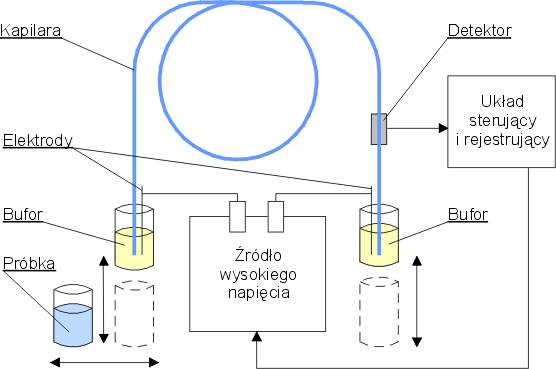
Rys. 1. Schemat układu pomiarowego
W skład układu do elektroforezy kapilarnej (rys. 1) wchodzą:
- Kapilary – najczęściej stosowane są kapilary ze stopionej krzemionki, pokryte poliimidową warstwą ochronną, o średnicy wewnętrznej 10-100 µm (350-400 µm średnica zewnętrzna) i długości od 20 do 100 cm. Końce kapilary umieszczone są w pojemnikach (o objętości kilku ml) wypełnionych odpowiednim buforem. Kapilara termostatowana jest za pomocą obiegu powietrza lub płynu chłodzącego.
- Zasilacze wysokiego napięcia – wysokie napięcie rzędu 5-30 kV przykładane jest do końców kapilary za pomocą dwóch elektrod platynowych umieszczonych w pojemnikach z buforem.
- System do wprowadzania próbek
- Detektor
- System sterowania układem i zbierania danych.
Całkowita objętość stosowanych kapilar wynosi kilka mikrolitrów. Aby uniknąć przeładowania, długość strefy próbki nie może być większa niż 1-2% całkowitej długości kapilary, co odpowiada objętości 1-50 nl. Wprowadzanie tak małych objętości wymaga specjalnego postępowania. Najczęściej stosowany jest sposób hydrodynamiczny, wykorzystujący różnice ciśnień między końcami kapilary, i elektrokinetyczny. Wlot kapilary umieszczany jest w pojemniku z próbką, a następnie przyłożenie ciśnienia u wlotu kapilary lub próżni u jej wylotu powoduje wprowadzenie próbki o objętości zależnej od czasu i różnicy ciśnień (rys. 2a i rys. 2b). Jeżeli pojemnik z próbką i zanurzonym wlotem kapilary umieszczony zostanie wyżej niż wylot kapilary, to próbka zostanie wprowadzona do kapilary dzięki przepływowi wynikającemu z różnicy poziomów (rys. 2c). Przy wprowadzaniu elektrokinetycznym w pojemniku z próbką oprócz kapilary umieszczana jest także elektroda. Po przyłożeniu stosunkowo niewielkiego napięcia (5-10 kV) składniki próbki migrują do kapilary dzięki swojej ruchliwości elektroforetycznej i przepływowi elektroosmotycznemu (rys. 2d).
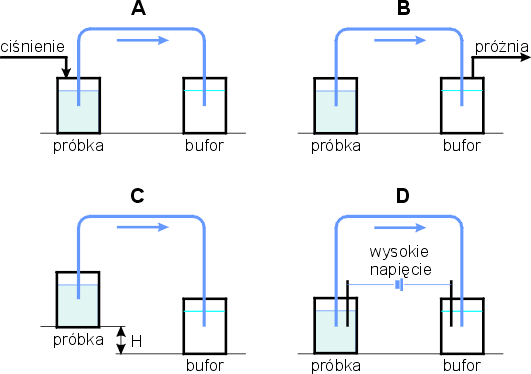
Rys. 2. Sposoby wprowadzania próbki do kapilary:
A, B, C – hydrodynamiczny, D-elektrokinetyczny
CO DZIEJE SIĘ W ŚRODKU KAPILARY
Podstawą rozdziału w elektroforezie kapilarnej są różnice w ruchliwościach elektroforetycznych oznaczanych analitów, co powoduje, że jony w polu elektrycznym poruszają się z różną prędkością ve:
ve = µeE
µe – ruchliwość elektroforetyczna
E – natężenie pola elektrycznego, które jest funkcją przyłożonego napięcia i długości kapilary.
Ruchliwość elektroforetyczna w danym roztworze jest wielkością charakterystyczną dla danego jonu i zależy od jego ładunku, promienia i właściwości buforu (pH, lepkości i siły jonowej oraz składu):
![]()
Dobierając odpowiedni skład buforu, możliwe jest kontrolowanie selektywności rozdzielania przez zmianę ruchliwości analitów. Wzrost stężenia i siły jonowej buforu powoduje zmniejszenie ruchliwości jonów, a dodatek organicznych rozpuszczalników (np. metanolu) zmienia lepkość roztworu. Istotny wpływ na ruchliwość jonów ma dodatek do buforu związków kompleksujących, a zastosowanie selektorów chiralnych (np. cyklodekstryn) umożliwia rozdzielenie enancjomerów. Zmiana pH buforu wpływa również na postać analitów, będących słabymi kwasami lub zasadami. Oprócz elektroforezy w kapilarze zachodzi inne zjawisko elektrokinetyczne, elektroosmoza – ruch cieczy wypełniającej kapilarę. W zetknięciu z roztworem, na skutek jonizacji grup silanolowych -Si-OH, wewnętrzna ściana kapilary jest naładowana ujemnie (rys. 3). Na zetknięciu fazy stałej i roztworu powstaje podwójna warstwa elektryczna o pewnym potencjale elektrokinetycznym ![]() . Składa się ona z części bezpośrednio przylegającej do ściany kapilary i części dyfuzyjnej, rozciągającej się w głąb roztworu o nadmiarowym ładunku dodatnim. Jeżeli taki układ znajdzie się w polu elektrycznym, to hydratowane kationy części dyfuzyjnej warstwy podwójnej poruszają się w kierunku katody, powodując przepływ cieczy w kapilarze. Przepływ elektroosmotyczny (EOF) opisuje wzór:
. Składa się ona z części bezpośrednio przylegającej do ściany kapilary i części dyfuzyjnej, rozciągającej się w głąb roztworu o nadmiarowym ładunku dodatnim. Jeżeli taki układ znajdzie się w polu elektrycznym, to hydratowane kationy części dyfuzyjnej warstwy podwójnej poruszają się w kierunku katody, powodując przepływ cieczy w kapilarze. Przepływ elektroosmotyczny (EOF) opisuje wzór:
![]()
| - stała dielektryczna; | |
| - potencjał elektrokinetyczny; | |
 |
- lepkość; |
| µEOF | - ruchliwość elektroosmotyczna; |
| E | - natężenie pola elektrycznego. |
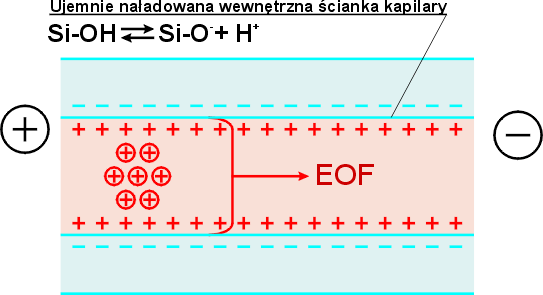
Rys. 3. Przepływ elektroosmotyczny
Przepływ elektroosmotyczny zależy od wielu parametrów, za pomocą których możemy kontrolować jego wielkość i kierunek:
- Zgodnie z równaniem zależy on proporcjonalnie od natężenia pola elektrycznego E. Jednak zbyt małe E może spowodować pogorszenie sprawności, natomiast zbyt duże E wpływa na wzrost niekorzystnego efektu generowania ciepła Joule’a;
- Ze wzrostem pH buforu wzrasta jonizacja grup silanolowych, co powoduje wzrost ładunku ujemnego na ściankach kapilary i wzrost EOF. Zmiana pH jest najprostszym sposobem doboru przepływu elektroosmotycznego.
- Zmiana stężenia lub siły jonowej buforu wpływa na potencjał elektrokinetyczny. Wzrost tych parametrów powoduje spadek potencjału elektrokinetycznego i zmniejszenie EOF. Jednak zbyt duże stężenie buforu jest niekorzystne ze względu na wzrost ciepła Joule’a.
- Dodatek organicznego rozpuszczalnika do buforu wpływa na zmianę potencjału elektrokinetycznego i zmienia jego lepkość, co zwykle powoduje zmniejszenie EOF.
- Dodatek kationowych związków powierzchniowo-czynnych do buforu, na skutek ich oddziaływania ze ścianami kapilary, powoduje zmniejszenie, a nawet odwrócenie kierunku przepływu elektroosmotycznego.
- Modyfikacja wewnętrznej powierzchni ścian kapilary powoduje eliminację lub zmniejszenie EOF.
Optymalizację szybkości przepływu elektroosmotycznego przeprowadza się w celu osiągnięcia odpowiedniej rozdzielczości w jak najkrótszym czasie, natomiast jego stabilność w czasie jest koniecznym warunkiem uzyskania powtarzalnych wyników. Cechą charakterystyczną przepływu elektroosmotycznego jest jego płaski profil (rys. 4), dzięki czemu metoda CE osiąga sprawności mierzone ilością półek teoretycznych rzędu 105 – 106. Dla porównania sprawności uzyskiwane dla HPLC wynoszą 30 – 100 tysięcy, dla GC 100 – 200 tysięcy półek teoretycznych.

Rys. 4. Profil przepływu hydrodynamicznego (A) i elektroosmotycznego (B)
Dzięki występowaniu EOF możliwe jest jednoczesne oznaczanie analitów dodatnich i ujemnych. Jeżeli przepływ elektroosmotyczny w kierunku od anody do katody jest większy niż prędkość elektroforetyczna anionów obecnych we wprowadzonej próbce, to zarówno kationy, aniony jak i indywidua obojętne będą poruszały się w kierunku katody (rys. 5). Do detektora najpierw docierają kationy, następnie nie rozdzielone, poruszające się z prędkością EOF indywidua obojętne, a na końcu aniony, przyciągane przez anodę w kierunku przeciwnym.

Rys. 5. Migracja jonów w kapilarze
W zależności od warunków, w jakich prowadzony jest proces rozdzielania, wyróżnia się różne typy elektroforezy kapilarnej:
- Strefowa elektroforeza kapilarna (CZE). Proces rozdzielania prowadzony jest w kapilarze wypełnionej w całej objętości tym samym buforem. Po przyłożeniu wysokiego napięcia składniki próbki obdarzone ładunkiem poruszają się z różną prędkością (rys. 6a).
- Micelarna elektrokinetyczna chromatografia kapilarna (MEKC). W procesie rozdzielania stosuje się bufory z dodatkiem związków powierzchniowo-czynnych, o stężeniu powyżej którego tworzą się micele. Składniki próbki, zarówno obdarzone ładunkiem, jak i neutralne, mogą oddziaływać z fazą micelarną, co pozwala polepszyć rozdzielenie składników jonowych i umożliwia rozdzielenie składników neutralnych (rys. 6b).
- Żelowa elektroforeza kapilarna (GCE). Rozdzielanie prowadzone jest w kapilarach wypełnionych żelami o odpowiednich średnicach porów, które działają jak sita molekularne. Składniki próbki migrują wzdłuż kapilary z szybkością zależną od ich wielkości.
- Izotachoforeza kapilarna (CITP). Rozdzielanie prowadzone jest z zastosowaniem niejednorodnego elektrolitu. Próbka wprowadzana jest do kapilary między dwa bufory, “wiodący” o ruchliwości jonu większej niż oznaczane jony próbki i “kończący” o ruchliwości jonu mniejszej niż jony próbki. Po przyłożeniu napięcia rozdzielane jony ustawiają się za buforem “wiodącym” według malejącej ruchliwości i migrują w postaci sąsiadujących ze sobą stref z prędkością jonów buforu “wiodącego” (rys. 6c).
- Izoelektryczne ogniskowanie kapilarne (CIEF). Jako podstawę rozdzielania wykorzystywane są różnice punktów izoelektrycznych (pl) oznaczanych analitów. W kapilarze generowany jest gradient pH (od najniższego przy wlocie do najwyższego przy wylocie kapilary). Po przyłożeniu napięcia składniki próbki poruszają się w kapilarze do momentu osiągnięcia strefy pH równego ich pl (rys. 6d).

Rys. 6.
A – Strefowa elektroforeza kapilarna (CZE)
B – micelarna elektrokinetyczna chromatografia kapilarna (MEKC)
C – izotachoforeza kapilarna (CITP)
D – izoelektryczne ogniskowanie kapilarne (CIEF)
PARAMETRY ANALITYCZNE OPISUJĄCE ROZDZIELENIE
W przypadku prowadzenia oznaczeń metodą CZE, MEKC oraz GCE poszczególne składniki próbki poruszają się w kapilarze wypełnionej jednorodnym buforem z różną prędkością w postaci wąskich stref. Czas, w jakim oznaczany składnik próbki dociera do detektora, nazywany jest czasem migracji:
tm=I L / µV
gdzie:
µ = µe + µEOF
I – efektywna długość kapilary (do detektora)
L – całkowita długość kapilary
Sygnały dla poszczególnych składników próbki rejestrowane są w postaci pików, a otrzymany elektroferogram pozwala na jakościowe i ilościowe oznaczenia. Sprawność rozdzielania podobnie jak w chromatografii opisywana jest liczbą półek teoretycznych:
N=µV / 2D
i w idealnych warunkach zależy od współczynnika dyfuzji D i przyłożonego napięcia. Dodatkowe rozmycie strefy próbki, pogarszające sprawność rozdzielania, może by spowodowane:
- Gradientem temperatury w kapilarze, spowodowanym nieefektywnym odprowadzaniem ciepła Joule’a;
- Zbyt dużą objętości wprowadzonej próbki;
- Adsorpcją składników próbki na ścianach kapilary
- Elektrodyspersją, spowodowaną różnicami w przewodnictwie strefy próbki i buforu;
- Przepływem laminarnym, kiedy poziom roztworów w obu pojemnikach z buforem nie jest jednakowy.
DETEKTORY STOSOWANE W CE
Ze względu na konieczność stosowania kapilar o bardzo małych średnicach i możliwości wprowadzania próbek o objętościach rzędu nl bardzo istotny jest dobór odpowiedniej metody detekcji (tabela).
| Metoda detekcji | Granice wykrywalności |
|---|---|
| UV | 10-5 – 10-8 |
| Fluorescencyjna ze wzbudzeniem laserowym |
10-7 – 10-9 10-8 – 10-12 |
| Elektrochemiczna amperometryczna konduktometryczna |
10-6 – 10-8 10-7 – 10-8 |
| Spektrometria mas | 10-8 – 10-9 |
Najczęściej stosowana jest detekcja UV. Wynika to z dużej uniwersalności tej metody, jak również ze względów technicznych. Kapilara, po usunięciu warstwy ochronnej, jest bezpośrednio umieszczona w celce pomiarowej detektora. Z powodu bardzo krótkiej drogi optycznej, równej średnicy kapilar, metoda ta należy jednak do najmniej czułych. Nowoczesne aparaty mogą być wyposażone również w detektory UV z przemiataniem widma, pozwalające na jednoczesne pomiary przy różnych długościach fal. Jednym z rozwiązań, umożliwiających polepszenie czułości, jest zastosowanie detektorów UV o zmienionej geometrii celki pomiarowej. Bardziej czuła jest metoda fluoroscencyjna, szczególnie z zastosowaniem lasera jako źródła wzbudzenia. Ponieważ ten rodzaj detekcji można zastosować tylko do niektórych związków, wymagana jest derywatyzacja próbki. Pozostałe detektory wymagają zastosowania specjalnych celek pomiarowych, do których wprowadzany jest koniec kapilary. W oznaczeniach związków elektroaktywnych obiecujące możliwości daje zastosowanie detekcji elektrochemicznej. W odróżnieniu od metod optycznych, mała objętość celki pomiarowej sprzyja osiągnięciu lepszej czułości. Trudności zastosowania detekcji elektrochemicznej to konieczność wyeliminowania wpływu wysokiego napięcia na układ pomiarowy detektora. Dużym osiągnięciem ostatnich lat było połączenie elektroforezy kapilarnej ze spektrometrią masową, umożliwiającą nie tylko ilościowe oznaczenia, ale także identyfikację oznaczanych analitów.
FIZYCZNE PARAMETRY ANALIZY
Dobór parametrów analizy jest niezwykle istotny dla jakości uzyskanych rozdziałów, a ich ilość znaczna. Poniżej podana jest ich lista – odnośniki prowadzą do ilustracji pokazujących ich wpływ na uzyskiwane elektroferogramy.
- Dobór kapilary
- Napięcie analizy
- Temperatura kapilary
- Dobór buforu
- Parametry wprowadzenia próbki
- Parametry detekcji
Wykorzystanie wszystkich technik elektroforezy kapilarnej pozwala na zastosowanie jej do oznaczeń wielu klas związków zarówno organicznych, jak i nieorganicznych. Stosowana jest w oznaczeniach aminokwasów, peptydów, białek, nukleotydów, DNA, węglowodanów, amin i witamin w próbkach biologicznych i płynach ustrojowych w analizie biochemicznej i biomedycznej. Istotne zastosowanie znalazła w farmacji w analizie czystości leków i do rozdzielania izomerów optycznych oraz w badaniach farmakokinetycznych. W ochronie środowiska metoda jest najczęściej stosowana do oznaczeń fenoli, pestycydów i herbicydów.
Wysoko sprawna elektroforeza kapilarna to metoda stale rozwijająca się. W naukowych czasopismach poświęconych tej metodzie prezentowane są wciąż nowe zastosowania zarówno do oznaczeń, jak i do badań właściwości fizykochemicznych analitów. Wiele prac poświęconych jest badaniom nad sposobami polepszenia czułości. Dotyczą one opracowywania nowych metod detekcji i zatężania próbki w kapilarze. Nowe możliwości rozdzielania daje zastosowanie kolumn pokrywanych.
- Główne zalety elektroforezy kapilarnej
- Niezwykle wysokie sprawności;
- Małe zużycie odczynników i rozpuszczalników organicznych;
- Małe objętości próbek;
- Krótkie czasy analiz;
- Możliwość prowadzenia pomiarów w szerokim zakresie pH;
- Możliwość jednoczesnego oznaczania kationów i anionów;
- Niskie koszty;
- Małe objętości powstających ścieków;
- Szerokie zastosowanie.